Genome-scale model reconstruction protocol
Reconstruction workflow overview
We reconstructed a cell-scale model of R. solanacearum that encompassed three modules, a genome-scale metabolic network, a macromolecule secretion module, and a virulence regulatory network. The two first modules are biochemical reaction networks whereas the regulatory network is a biochemical interaction network. Thus, the modular structure of this resulting ‘hybrid model’ (Le Novere et al. 2015) allows computational approaches with appropriate methods for both types of networks, i.e. constraint based modeling for the biochemical networks and multi-state logical modeling for the regulatory network. Both methods do not require kinetics parameters and thus are relevant for genome-scale analysis. We took advantage of our recently developed application to perform computation of such hybrid model (Marmiesse et al. 2015).

Overview of the hybrid model reconstruction and modelling
Metabolic model reconstruction
Pipeline Overview
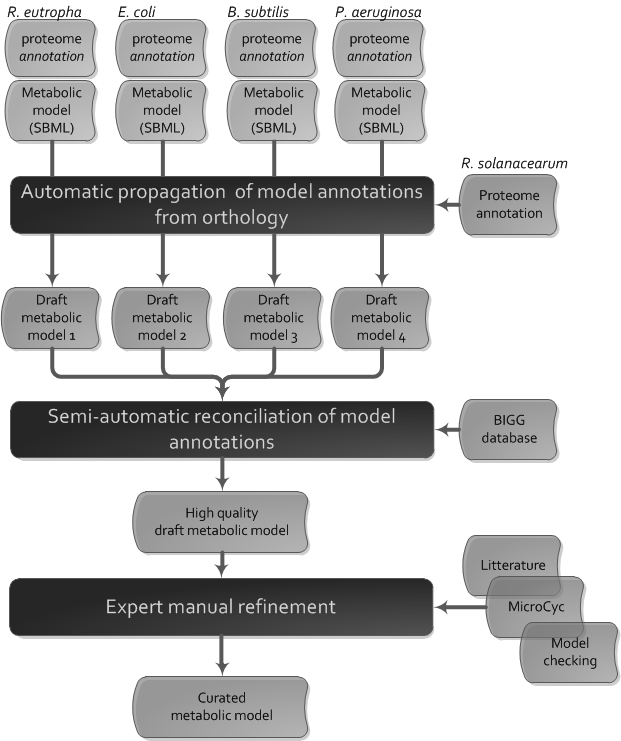
Overview of the metabolic network reconstruction pipeline
The reconstruction of the metabolic model of R. solanacearum has been performed in four steps:
- Reconstruction of several draft metabolic models from curated metabolic models of other species based on orthology
- Semi-automatic standardisation of the draft metabolic models
- Semi-automatic merging of the draft metabolic models
- Expert curation of the metabolic model by browsing litterature, Microcyc and by performing model-checking.
Automatic propagation of model annotations from orthology
Draft metabolic models have been built from these four published metabolic models: Ralstonia eutropha (RehMBEL1391) (Park et al, 2011)) Bacillus subtilis (Bs_iYO844 (Oh et al, 2007)), Pseudomonas aeruginosa (iMO1086 (Oberhardt et al, 2011)) Escherichia coli (iJO1366 (Orth et al, 2011)) The two first ones were selected because of their phylogenetic proximity with R. solanacearum. The bacterium P. aeruginosa was selected because of its pathogen lifestyle and the model of E. coli was used because of the high quality of its reconstruction. The Systems Biology Markup Language (SBML) is for years the standard file format to exchange metabolic models (Hucka et al, 2003). We naturally use this format for all our reconstruction and analysis steps. Three metabolic models (B. subtilis, P. aeruginosa, and E. coli) were published in this format and so easily collected while the model of R. eutropha had to be converted from pdf to SBML since pdf was the only format supplied by the authors for their model. We downloaded proteins sequences of the entire genome of the sources organisms from NCBI, plus the one of R. solanacearum strain GMI1000, later called the “target organism”, from the official genome web portal1 (Salanoubat et al, 2002).
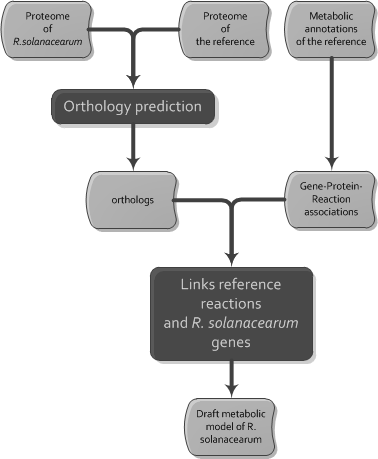
Propagation by orthology of the gene reaction association from a reference model to a draft model (inspired by the Autograph method (Notebaart et al, 2006))
We used the Autograph method (Notebaart et al, 2006) to automatically transfer by orthology the gene reaction associations of the four reference models in four draft models (Figure 2). The orthology prediction was made by using a tuned version of Inparanoid (Remm et al, 2001). Inparanoid found orthologs between the proteome of R. solanacearum and each of the four reference proteomes. The Blast results obtained in Inparanoid were filtered by only selecting hits for which the identity exceeds 30 % and the coverage exceeds 50 %. The BLOSUM45 matrix (reasonable for prokaryotes) was used for the Inparanoid bootstrapping step. For the propagation step, we developed a function called PropagateGprsFromOrthologs in the parseBioNet package (JAVA package dedicated to metabolic networks that we develop for some years). PropagateGprsFromOrthologs takes as input the SBML reference file and a tabulated file containing for each protein of R. solanacearum the ortholog in reference specie, and returns a SBML file. Reactions in the reference model are included by PropagateGprsFromOrthologs in the target model if at least one reference gene has an ortholog in R. solanacearum. In the gene protein reaction links of the kept reactions, the identifiers of the genes are replaced by the identifiers of the corresponding orthologs in R. solanacearum or by the mention “no_ortholog” when the gene does not have any identified ortholog in R. solanacearum. Reactions without associated genes are propagated. Results of the automatic propagation step are displayed in Figure 3. The proportion of reactions propagated from each reference model is quite high (> 60%). We will see in the next section that the manual curation performed after merging highly reduces this number.

Propagation results. Number and type of reactions propagated from each reference model. Click on the circles to change the layout.
Semi-automatic standardisation of the draft metabolic models
Since the reference models have been made by different teams, they use different ontologies for their reaction and metabolite identifiers. For instance, the identifier of the adenosine triphosphate is M_atp_c in the models of B.subtilis, E. coli and R. eutropha while it’s C0002 in the model of P. aeruginosa. So, before merging the four draft models, we needed to standardize the identifiers of their metabolites and reactions. Automatic reconciliation tools have been recently described (Bernard et al, 2012). However, they have some drawbacks. First, they often rely on information (Inchi, SMILES, cross-references) found in public metabolic databases to reconciliate reaction and metabolite identifiers. Secondly, the process is always fully automatic and does not offer ways to complete or correct the produced dictionary. These two drawbacks are explained by the fact that these methods have been first essentially designed to quickly reconciliate complete metabolic databases, such as KEGG (Kanehisa & Goto, 2000), MetaCyc (Caspi et al, 2014) or BIGG. (Schellenberger et al, 2010). For our topic, we wanted to quickly and without ambiguities reconciliate the identifiers of the four draft metabolic models produced by the propagation step. For this, we developed a web-designed semi automatic tool called SAMIR (Semi Automatic Metabolic Identifier Reconciliation). SAMIR is generic since the standard identifiers (as well as the identifiers to standardize) are provided by a SBML file (the standard format for metabolic networks (Hucka et al, 2003)) that can be built from public or home-made metabolic database. In the same way than MNXref (Bernard et al, 2012), the process of the reconciliation of metabolites use the reaction context. If two reactions are identified as identical in the two SBML files, then mapping between the metabolites involved in these reactions is proposed. In the same way, if two reactions involve metabolites identified as identical, they will be proposed for the reconciliation. The main difference between MNXRef and SAMIR is that the former tries to validate the potential mappings and iterates until no new mappings can be obtained while the latter stops after each iteration and let the user to decide which mapping is the best. The second alternative is certainly more time consuming but has the great advantage to obtain an on-the-fly curated reconciliation at the end. A system of score has been designed to help the user to decide between several mappings and the potential mappings are sorted by their score in the interface (.a). For both metabolites and reactions, the score includes a measure of the name similarity. For reactions, the score includes also the percentage of metabolites identified as identical and a bonus if the two EC numbers are identical. For metabolites, the score includes bonus if the chemical formulae (potentially provided with the SBML file) are the same or if the pair to check is the last one not mapped in a pair of identical reactions. Matching entities and ranking of probable matches are listed in the web interface allowing annotators to curate matches.The web interface allows to declare some a priori on the identical identifiers (provided for instance by the methods cited above), on the identifiers that are definitely different (and that won’t be proposed for the reconciliation) and on the identifiers that are unique to the network to standardize and that can be added at the end to the reference SBML file. To our knowledge, SAMIR is moreover the first reconciliation method which takes into account compartments in the models by declaring which compartments are identical in the two SBML files. After each iteration, a pie chart indicates to the user the progression of the reconciliation (.b) and displays the links to the standardized SBML file, the produced dictionary and the reference SBML file completed with the reactions unique to the SBML file to standardize. Since SAMIR is generic and publicly available in a Beta version1, it can be used to reconciliate other metabolic reconstructions.Semi-automatic reconciliation of the standardised draft metabolic models in one high-quality draft metabolic model
The resulting four propagated networks into the target organism are merged following an expert evaluation of the best reactions candidates propagated from the sources considering the completeness of the GPRs, ranking of blast results, and quality of the annotation in the source GEMs. We developed a parseBioNet application called CompareSbmls which returns information about reactions shared between models, gene protein reaction links shared between reactions to accelerate this step.
Venn diagram representing the intersection between each model built by propagation and the final model
We launched SAMIR to build four standardized metabolic models from the four draft models built by propagation. To be able to compare our model with most of the metabolic models, we chose the BIGG database for which the identifiers have been used to build most of the existing metabolic models. We can see in Figure 5 that only 287 propagated reactions are found in every reference model. Also, a lot of the reactions (515) propagated in the final model exclusively come from E. coli. This can easily be explained by the completeness of this model which is not found in the other ones. Numerous reactions have been manually removed from the propagation results (e.g 671 reactions from E.coli). Indeed, orthology evidence is often not sufficient to add a reaction in the model. The metabolic context, deduced by the other reactions and by the literature, thus made reactions dismissed. At last, 853 reactions which don’t come from the propagation results have been manually added to the final model . This highlights the importance of manual curation after a propagation step.